Algorithm
Overview
MembraneCurvature calculates mean and Gaussian curvature of surfaces derived from atoms of reference in 4 steps:
3. Derive surface and calculate surface derivatives
A summary of the algorithm used in MembraneCurvature is shown in the following diagram:
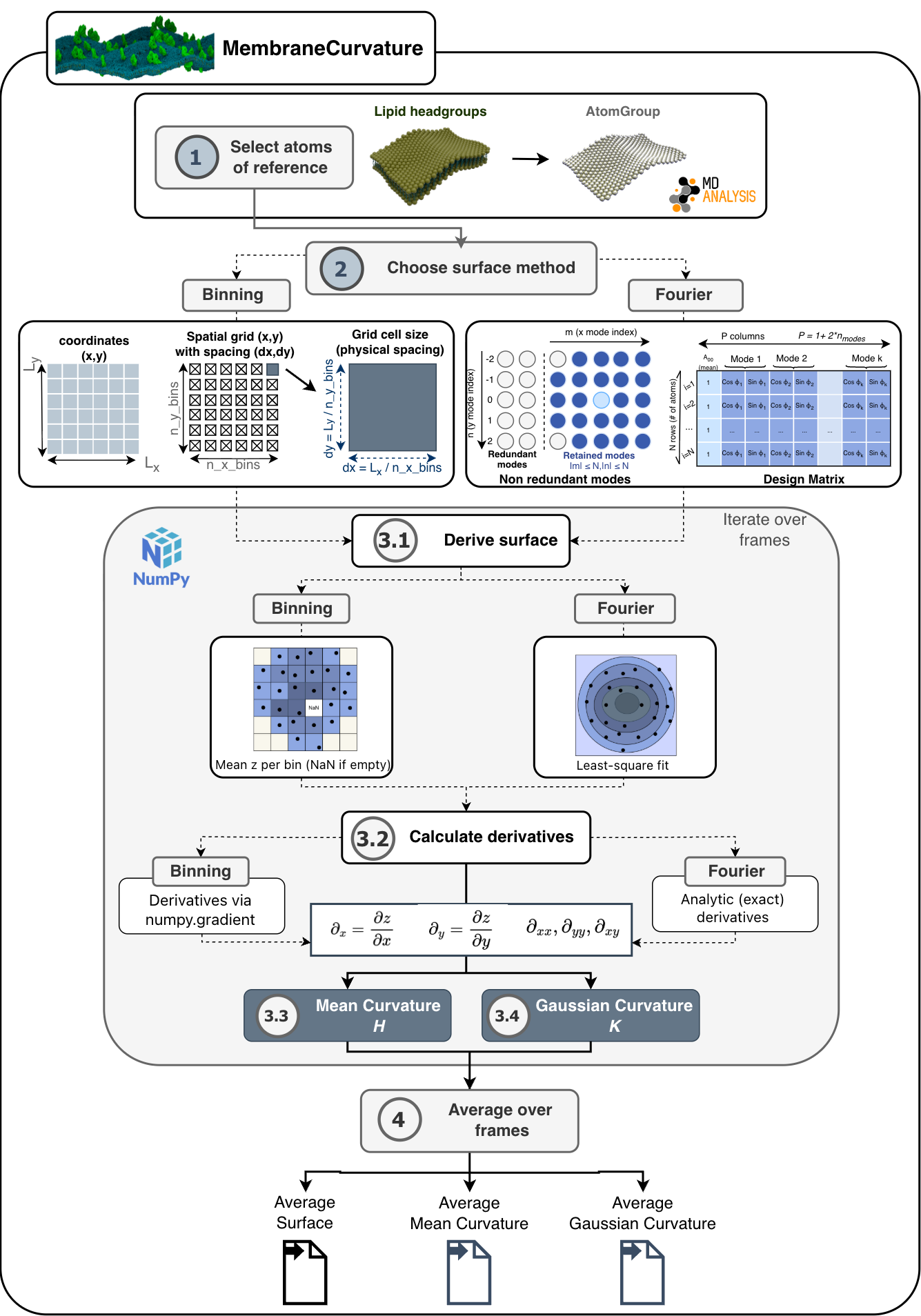
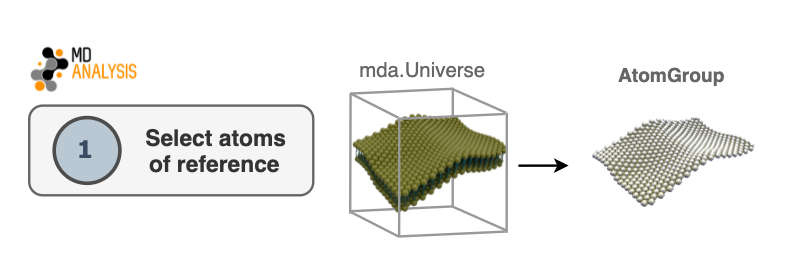
1. Select atoms of reference
The first step in the algorithm consists of selecting atoms that will be used as
a reference to derive a surface. This selection will be contained in an
AtomGroup. Typically in biological membranes,
lipid headgroups are the most common elements to use as an AtomGroup of
reference.
2. Choose surface method
Two surface-derivation methods are available, selected via the
surface_method argument of
MembraneCurvature.
2.1 Fourier method (
surface_method='fourier', default method)A truncated 2D Fourier series is fitted to atom heights by linear least squares at each frame. Partial derivatives are evaluated analytically from the fitted series on the same grid as the binning method, avoiding finite-difference discretization error. See
fourier_surfacefor details.2.2. Binning method (
surface_method='binning')Atoms are assigned to bins on a regular grid and the height of each bin is set to the mean \(z\)-coordinate of its atoms. Partial derivatives are estimated numerically from the discrete height field using
numpy.gradient()with the physical bin spacing. Seebinning_surfacefor details.
2.1 Fourier method
surface_method='fourier' is the default method used in Membrane Curvature.
The Fourier method fits a truncated periodic 2D Fourier series to atom heights
by linear least squares at each frame.
The truncation is controlled by fourier_m and fourier_n (default 2).
The basis is periodic on the simulation box with periods \(L_x\) and \(L_y\),
so the fitted surface is consistent with periodic boundary conditions in \(x\) and
\(y\).
The Fourier expansion used as a basis function is given by:
Here, \(k_x\) and \(k_y\) are the fundamental wavevector components:
therefore the phase for the mode (m,n) is
The Fourier method in MembraneCurvature is conceptually related to Fourier surface modeling [CAG2009] and molecular Fourier shape descriptors [JMG1988], but it is not a direct implementation of either paper. The least-squares height-field fit used in MembraneCurvature is tailored to the AtomGroup of reference given their cordinates.
Shen et al., Fourier method for large-scale surface modeling and registration, Computers & Graphics (2009), doi: 10.1016/j.cag.2009.03.002.
Leicester et al., Description of molecular surface shape using Fourier descriptors, Journal of Molecular Graphics (1988), doi: 10.1016/0263-7855(88)85008-2.
The full Fourier surface workflow comprises six steps, where the first four steps build and
solve a linear model that reconstructs the height field from plane-wave basis
functions. The final two steps evaluate the fitted surface and its derivatives
analytically on a grid of bin centres. In the following sections, we describe the
overall workflow implemented in MembraneCurvature. For details on each step and
the associated functions, see the API documentation in
fourier_surface.
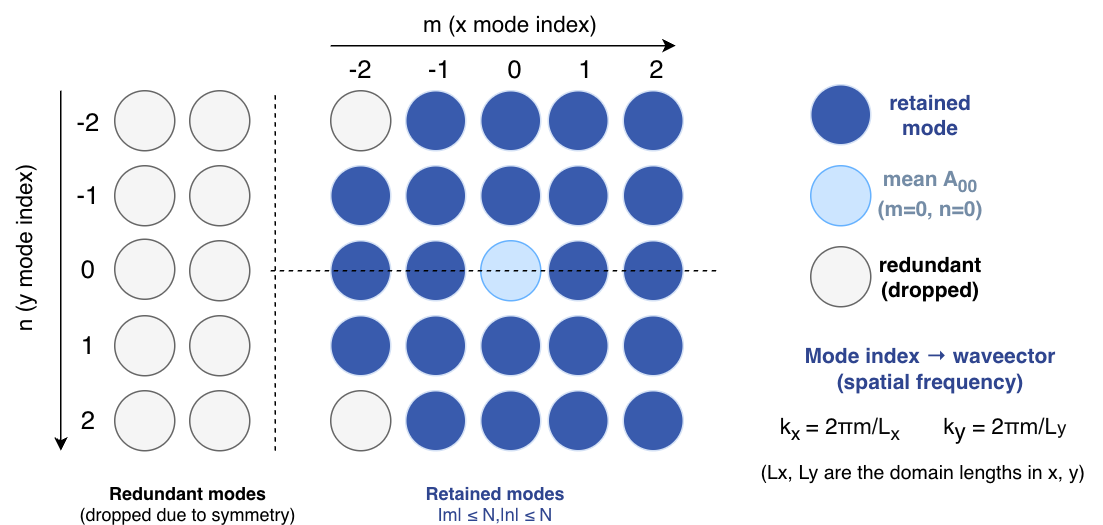
2.1.1 Choose Fourier modes
The first step is to choose a non-redundant set of Fourier modes with
fourier_mode_list() and determine the
total parameter count with n_fourier_parameters().
This removes conjugate redundancy for real-valued surfaces and isolates the
mean term.
2.1.2 Compute wavevectors
We then compute the wavevector components (\(k_x\), \(k_y\)) for each mode
using _compute_wavevector(). These
set the phase \(\phi = k_x x + k_y y\) that appears in each cosine/sine
basis function.
MembraneCurvature builds the non-redundant mode list via fourier_mode_list(M, N)()
and computes the total parameter count with n_fourier_parameters(M, N)().
MembraneCurvature then validates that the AtomGroup of reference
contains at least that many atoms and raises a ValueError if the selection is too small.
Warning
The explanation above is for the users to understand how MembraneCurvature builds the mode list and parameter count internally.
Do not pass the mode list fourier_mode_list(M, N)()
or parameter count n_fourier_parameters(M, N)()
directly. MembraneCurvature builds them internally.
Users choose the Fourier truncation via the constructor arguments fourier_m and fourier_n
By passing the maximum mode indices, MembraneCurvature builds the actual mode list and computes the total
parameter count.
Important
Because the derivatives are analytic, the Fourier method is not subject
to finite-difference discretization error from the bin grid. Curvature
still depends on the Fourier series truncation: use
fourier_m = fourier_n = 2 unless shorter wavelengths are required, and
increase these values only while curvature improves systematically rather than
becoming dominated by noise.
2.1.3 Build design matrix
We then build the design matrix with _build_fourier_matrix().
Each row of the design matrix corresponds to an atom position and columns are the constant offset
followed by \(\cos(\mathbf{k}\cdot\mathbf{r})\), \(\sin(\mathbf{k}\cdot\mathbf{r})\)
pairs for every retained mode. This matrix encodes the linear relation between the
Fourier coefficients and the observed heights.
The design matrix \(\mathbf{\Phi}\) is a matrix of shape \((N, P)\) where
\(N\) is the number of atoms in the AtomGroup
of reference, and \(P\) is the number of parameters. \(P\) is defined
as \(P = 1 + 2\,n_{\text{modes}}\) where \(n_{\text{modes}}\) is the number of
the k retained Fourier modes.
We can conceptualize \(\mathbf{\Phi}\) as a matrix with rows corresponding to atom positions and columns corresponding to the basis functions (cosine and sine of the wavevector \(k\)) evaluated at each atom position, so that the fitted heights satisfy the linear system of equations \(\mathbf{z} \approx \mathbf{\Phi}\,\boldsymbol{\theta}\), from which we can solve for the Fourier coefficients by solving a least-squares system.
A single row \(i\) of \(\mathbf{\Phi}\) for an atom at \(\mathbf{r}_i=(x_i,y_i)\) has the form:
With this formulation, we can solve for the Fourier coefficients by solving a least-squares system:
where \(\boldsymbol{\theta}\) is the vector of Fourier coefficients. These coefficients are the ones we use to reconstruct the height field and calculate the derivatives.
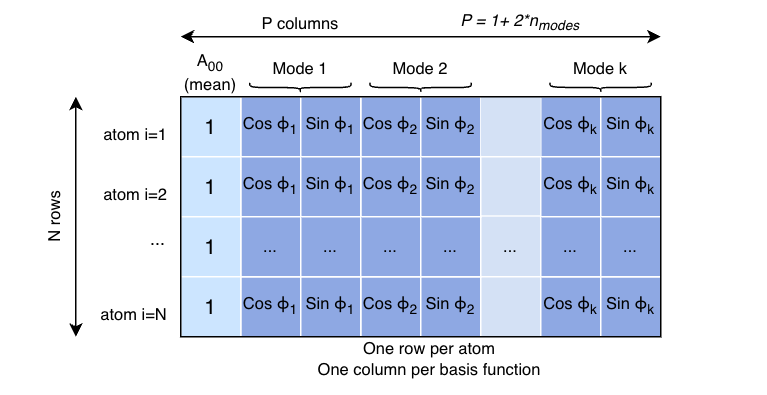
Note that the column ordering follows the mode list returned by
fourier_mode_list(). For each retained
mode (m,n) the design matrix contains a cosine column then a sine column,
so columns appear as 1, cos_{(m1,n1)}, sin_{(m1,n1)}, cos_{(m2,n2)}, sin_{(m2,n2)}, ....
Important
In summary:
The design matrix \(\mathbf{\Phi}\) has shape \((N, P)\) where rows (\(N\)) are atoms
and columns (\(P\)) are the basis functions. \(N\) is the number of atoms in the
AtomGroup of reference. \(P\) is the total number of parameters:
which is the sum of one constant column (the mean term \(A_{00}\)) plus two columns for each retained Fourier mode (one for cosine and one for sine).
2.1.4 Solve least-squares system
We then solve the least-squares system for the Fourier coefficients using
_fourier_fit_from_atoms(), which
calls _solve_design_least_squares_svd().
The latter function solves the linear least-squares system via truncated SVD.
This function solves the least-squares system for the Fourier coefficients and splits the solution
vector into the mean term \(A_{00}\) and the per-mode amplitudes \(A_{mn}\) and \(B_{mn}\)
with _unpack_coefficients().
The returned coefficients are then used to evaluate the fitted height and its
analytic derivatives on the bin-centre mesh via _eval_fourier_surface().
The Fourier coefficients are obtained by linear least squares. The coefficients are obtained by minimizing the residual sum of squares between the observed heights and the fitted heights:
via truncated SVD (_solve_design_least_squares_svd()).
Because the model is linear in
\(\boldsymbol{\theta}\), no nonlinear optimisation is required.
If the effective rank of \(\mathbf{\Phi}\) is smaller than \(P\),
a UserWarning is emitted. The truncated-SVD solver still returns a well-defined
minimum-norm least-squares solution, but the coefficients are not uniquely
determined by the data.
Overall, truncated SVD lets us fit the best surface even when the data can’t uniquely determine every Fourier coefficient, and it reduces noise amplification in modes that are not well-determined by the data.
2.2. Binning method
The binning method derives a surface by partitioning the simulation box into
a regular n_x_bins x n_y_bins grid along the \(x\) and
\(y\) directions. For each bin we compute the average \(z\) coordinate
of the atoms that fall inside the bin to form the discrete height field.
The resulting discrete height field is then differentiated numerically
using numpy.gradient() to obtain the partial derivatives required for
curvature calculation. Set surface_method='binning' explicitly to use
this method; it requires no additional parameters beyond the grid dimensions.
In the next section, we describe the details of the binning method.
2.2.1. Set grid
The dimensions of the grid are determined by the size of the simulation box
contained in the Universe.
The grid covers a rectangular domain in the membrane plane. By default that
domain matches the x and y edges of the simulation box from MDAnalysis’
Universe. The grid comprises n_x_bins x n_y_bins
bins along the x and y directions. Note that the user can define the number of
bins via the n_x_bins and n_y_bins arguments.
For every atom in the AtomGroup of reference,
MembraneCurvature assigns it to a grid cell based on its x and y coordinates.
In practice, each coordinate pair (x, y) is mapped to a grid location
[l, m] corresponding to a bin in the discretized membrane surface.
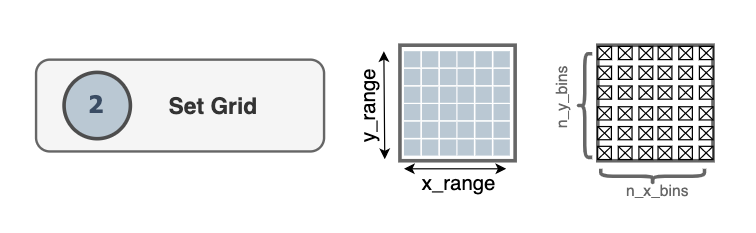
Here, \(L_x\) and \(L_y\) denote the size of the simulation box in the
x and y directions, respectively, while the size of the region covered by
the grid is represented by x_range and y_range, along the x and y
directions, respectively. The spacing between grid points in each direction is
then determined by dividing these lengths by the number of bins.
Note
Unless the user provides a different input, MembraneCurvature will determine
the dimensions of the grid based on the size of the box on the first frame via
dimensions.
grid_dimension_x = (0, universe.dimensions[0])
grid_dimension_y = (0, universe.dimensions[1])
2.2.2. Populate grid
Once the grid has been populated, the z coordinates of atoms assigned to each cell are collected to form a height field over the grid.
Coordinates are converted to integer bin indices via scale factors
We histogram the atoms of reference into a 2D grid by mapping each atom at \(x\) and \(y\) coordinates to an integer bin index. The scale factor converts coordinates from length units into bin units so that flooring yields an integer index:
and similarly for \(y\).
Atoms that map outside the valid bin range
(negative indices or indices ≥ n_x_bins/n_y_bins) are skipped and
trigger a one-time detailed warning. The function uses a WarnOnce helper so the full diagnostic
is shown on first occurrence and a short message on subsequent occurrences.
To populate the grid, MembraneCurvature wraps
the coordinates of the AtomGroup of reference
using wrap() only when wrap=True is
set (the default for surface_method='binning').
Empty bins (zero samples) are represented as numpy.nan in the returned
(n_x_bins, n_y_bins) array: the implementation replaces zero counts
with numpy.nan and divides summed z-values by the per-bin counts. As a result,
trajectory averages use numpy.nanmean() and therefore ignore empty bins.
Warning
The binning routine itself does not apply periodic wrapping;
MembraneCurvature applies
AtomGroup.wrap() only when wrap=True is set.
Set
wrap=Trueto pack atoms back into the grid if you are calculating curvature on a raw trajectory.Set
wrap=Falseto omit atoms from the simulation box that fall outside the grid when you are calculating curvature on:a trajectory (membrane only or membrane-protein with position restraints) that already pre-processed periodic boundary conditions.
a membrane-protein system that already pre-processes rotational and translational fit for the protein.
3. Derive surface and calculate surface derivatives
For every frame of the trajectory, the surface derived from the
AtomGroup is
calculated and stored in z_surface.
Similarly, the calculation of mean and Gaussian curvature is performed in every
frame and stored in MembraneCurvature.results.mean and
MembraneCurvature.results.gaussian, respectively.
The following sections describe the details of the two methods used to derive the surface and calculate its derivatives.
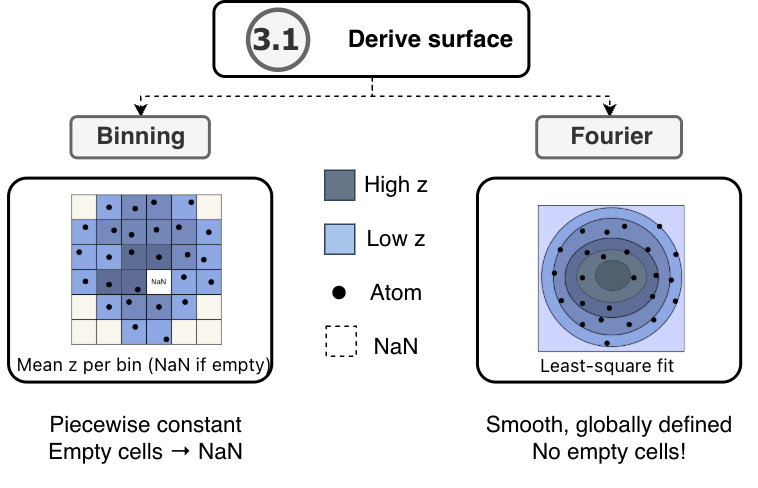
3.1. Derive surface
The surface is derived from atom positions using the selected method
and stored in z_surface for each frame.
With surface_method='binning', the height field is the discrete
\(N_x \times N_y\) array of per-cell mean \(z\) coordinates
assembled in 2.2.1. Set grid. Bins containing no atoms are set to NaN
and excluded from trajectory averages.
With surface_method='fourier', the height field is the fitted Fourier
series evaluated at bin centres after the least-squares fit described in
2.1 Fourier method. Because the representation is continuous and periodic
on the simulation box, every bin centre receives a value and no NaN
entries arise.
3.2. Calculate derivatives
Five partial derivatives of the height field are required by the curvature formulas: the first derivatives \(\partial_x\) and \(\partial_y\), the second derivatives \(\partial_{xx}\) and \(\partial_{yy}\), and the mixed derivative \(\partial_{xy}\).
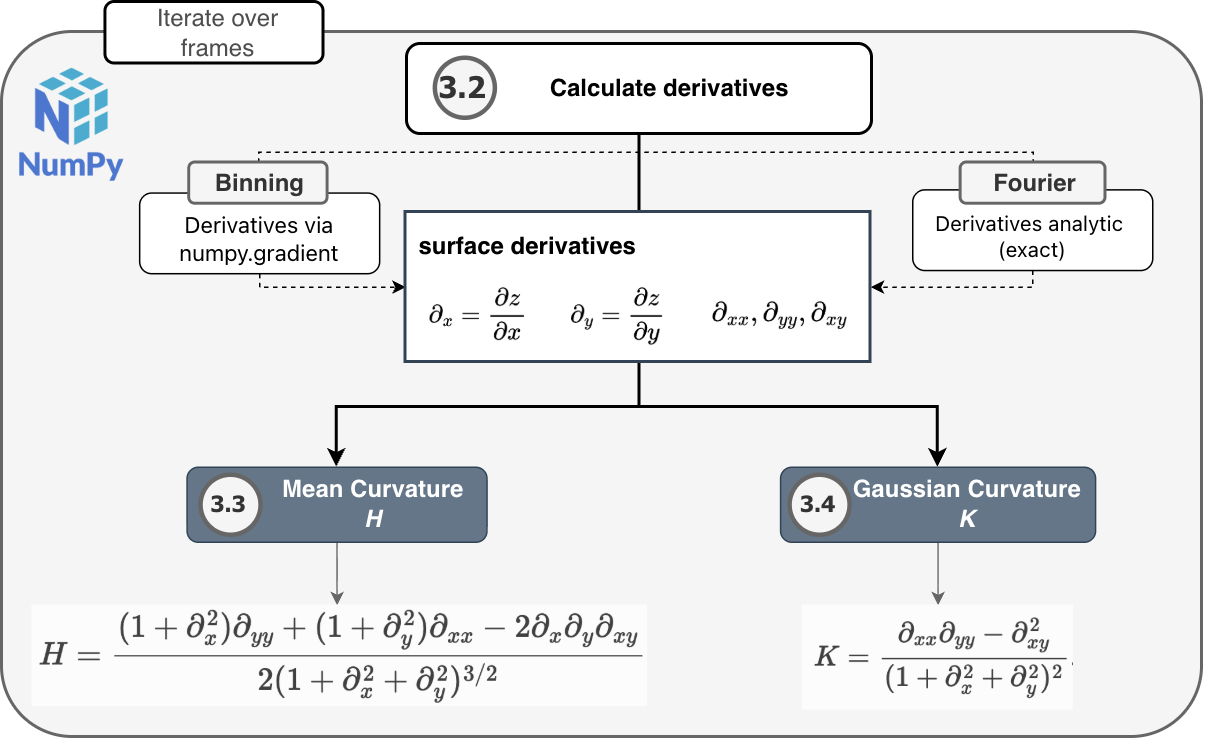
Important
There is a key difference between the binning and Fourier methods when it comes to calculating the derivatives!
With the binning method, the derivatives are estimated numerically from the discrete height field using
numpy.gradient()with the physical spacings \(\Delta x\) and \(\Delta y\), so that curvature values are in physical units.With the Fourier method, the derivatives are evaluated analytically from the fitted Fourier series, so they are not subject to finite-difference discretization error. Curvature accuracy is instead governed by the Fourier series truncation.
Therefore, the two methods differ in what limits curvature accuracy: binning is sensitive to finite-difference error on the height grid (often worse when the grid is coarse), while the Fourier pipeline is sensitive to how well the truncated series fits the atom data; its derivatives are exact for that fitted surface at any output grid resolution, so refining the bin grid alone does not remove truncation or sampling limitations.
3.2.1. Binning + finite differences (surface_method='binning')
With surface_method='binning', these are estimated numerically from
the discrete height field using numpy.gradient() with the physical
spacings \(\Delta x\) and \(\Delta y\), so that curvature values
are in physical units. Because this step uses finite differences, very
coarse grids may introduce discretization error.
Note
These derivatives are evaluated using the actual grid spacing (dx, dy),
so that changes in height are measured per unit distance in real space rather
than per grid index. This makes curvature values physically meaningful and
reduces their sensitivity to the chosen grid resolution.
Curvature is computed from surface derivatives evaluated using the grid spacing
(dx, dy), ensuring results are expressed in physical units and are less
sensitive to grid resolution. Because the derivatives are computed numerically,
very coarse grids may still affect curvature estimates due to finite-difference
discretization error.
For details on the binning method, see API documentation in
binning_surface that describes every
associated functions.
3.2.1.1. FFT filtering on the averaged surface (fft_filter)
A brick-wall filter is applied to the averaged surface once all frames have been processed
when surface_method='binning' and the argument fft_filter is set to 'auto' (default) or
passed as a dictionary like {'q': (q_low, q_high)}.
Important
The FFT filter is not applied to per-frame surfaces. Filtering is performed on the time-averaged height field, where thermal fluctuations have already been suppressed by averaging.
Pass-band limits are resolved at construction time via
resolve_fft_filter() and applied at the end
of the run with apply_fft_filter() (see
fft_filtering).
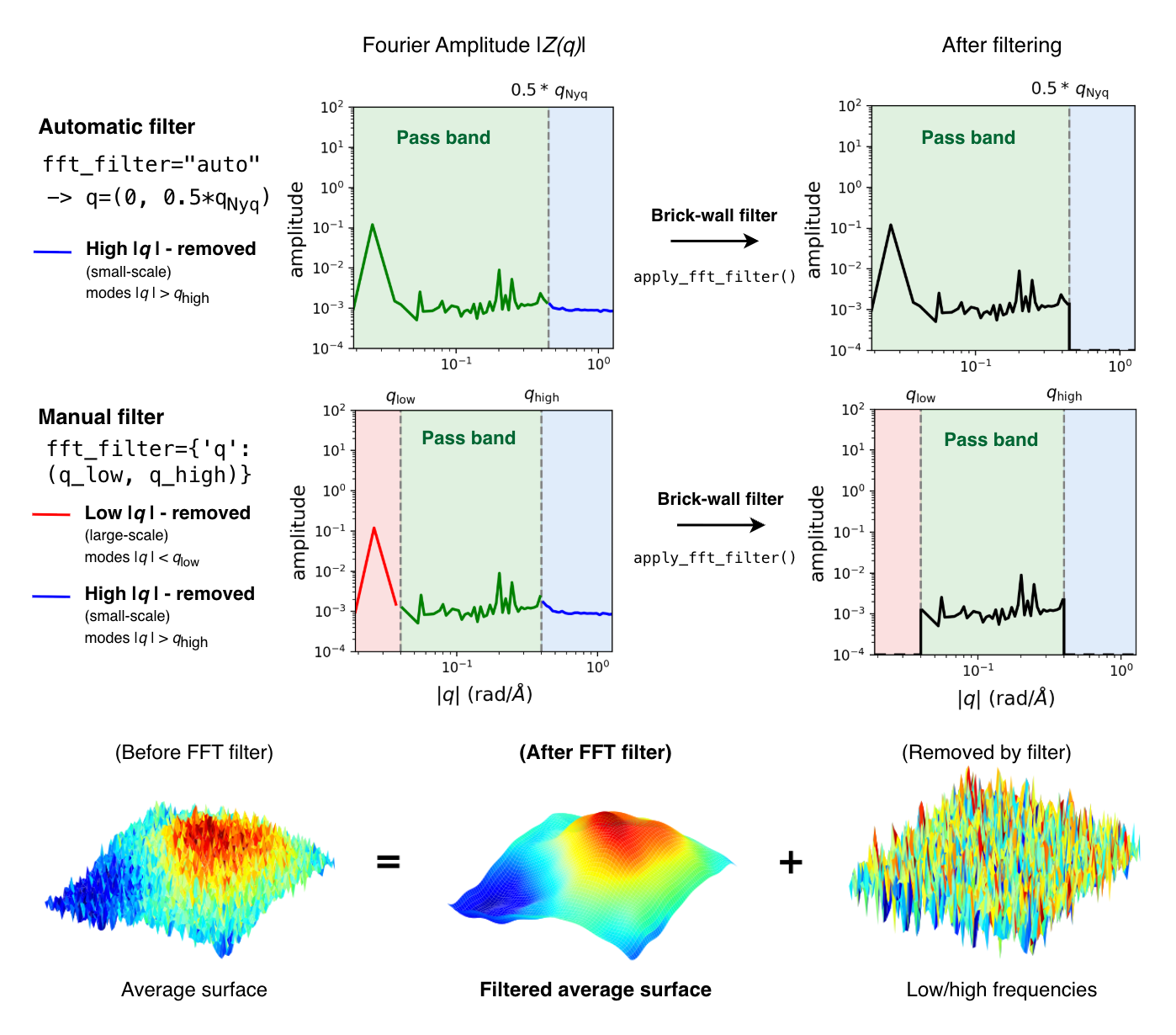
For filtered average surface maps, MembraneCurvature
averages the height field (z_surface)
over the trajectory and then smooths it in reciprocal space by zeroing all Fourier modes outside
the pass band defined by \(q_{\mathrm{low}} \leq |q| \leq q_{\mathrm{high}}\) via
apply_fft_filter() before transforming back to real space.
The resulting filtered surface is stored in average_z_surface,
and then used to calculate mean and Gaussian curvature via mean_curvature() and
gaussian_curvature(), respectively.
With the default fft_filter='auto', the pass band is \((0,\ 0.5\,q_{\mathrm{Nyq}})\),
a conservative low-pass by default which retains the large-scale membrane shape while
suppressing short-wavelength noise.
For binning, filtering defaults to fft_filter='auto'. To control the band manually,
pass fft_filter={'q': (q_low, q_high)} in rad/Å. To disable filtering altogether, pass fft_filter=None.
Warning
Before the FFT filtering, empty bins are temporarily filled with the mean height of occupied
bins, then restored to NaN after the inverse FFT. Large empty regions can
introduce broadband spectral contamination and distort the filtered surface near gaps.
Prefer denser binning or smaller empty regions when filtering is enabled.
Note
The pass-band mask is isotropic in \(|q|\). For non-square bins
(\(\Delta x \neq \Delta y\)), modes that are resolvable along the finer axis
but exceed \(q_{\mathrm{Nyq}} = \min(\pi/\Delta x,\, \pi/\Delta y)\) are
removed. Since numpy.fft.fft2() assumes a periodic grid, use wrap=True,
as recommended for binning in general.
3.2.2. Fourier fit + analytic derivatives (surface_method='fourier')
With surface_method='fourier', the partial derivatives are evaluated
analytically from the fitted Fourier series (see 2.1 Fourier method),
so they are not subject to finite-difference discretization error.
Curvature accuracy is instead governed by the Fourier series truncation.
Warning
Use fourier_m = fourier_n = 2 unless shorter wavelengths are needed, and
increase the mode indices only while curvature improves systematically.
Note
Because no finite-difference step is involved, the analytic derivatives are exact with respect to the fitted surface, regardless of grid resolution.
For details on the Fourier method, see API documentation in
fourier_surface that describes every step with its
associated functions.
3.3. Mean curvature
Mean curvature \(H\) is calculated from the five partial derivative arrays using the Monge-gauge formula:
The result has units Å -1 and is stored in
MembraneCurvature.results.mean for each frame.
3.4. Gaussian curvature
Gaussian curvature \(K\) is calculated from the same derivative arrays using:
via gaussian_curvature_monge().
As for the calculation of mean curvature, Gaussian curvature is calculated for
every frame and the result has units Å -2 and is stored in
MembraneCurvature.results.gaussian for each frame.
Warning
The Monge-gauge formulas in steps 3.3 and 3.4 are exact. There is no small-gradient approximation applied. Both methods feed the same five derivative arrays into the same formulas; the only difference is how those derivatives were obtained in step 3.2. Calculate derivatives.
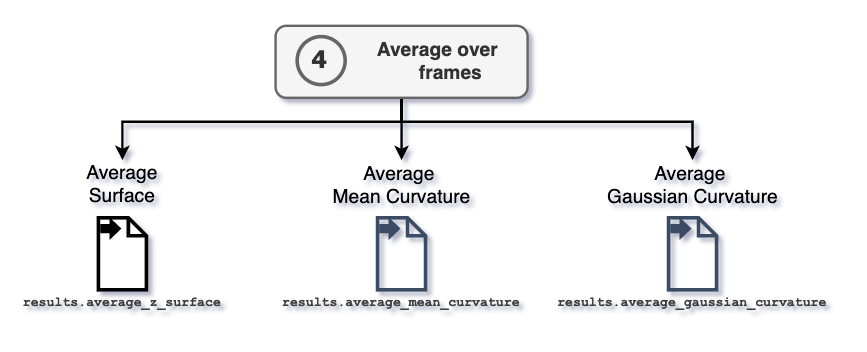
4. Average over frames
The attributes MembraneCurvature.results.average_mean and
MembraneCurvature.results.average_gaussian contain the computed
values of mean and Gaussian curvature averaged over all the
n_frames in the trajectory.
After performing the average over frames, the information of average surface,
mean, and Gaussian curvature are stored in the
MembraneCurvature.results.average_z_surface,
MembraneCurvature.results.average_mean, and
MembraneCurvature.results.average_gaussian arrays, respectively.
Each array has shape (n_x_bins, n_y_bins).